THALASSEMIA ALPHA AND BETA
Thalassemia is one of genetic blood diseases which vary widely in severity. Patient with Thalassemia disease are not able to make enough hemoglobin, which causes severe anemia. Hemoglobin is found in red blood cells and carries oxygen to all parts of the body.
When there is not enough hemoglobin in the red blood cells, oxygen cannot get to all parts of the body. Organs then become starved for oxygen and are unable to function properly.

Hemoglobin contains two different kinds of protein chains named alpha and beta chains. Any deficiency in these chains causes abnormalities in the formation, size, and shape of red blood cells.
People whose hemoglobin does not produce enough alpha protein have alpha thalassemia. It is commonly found in Africa, the Middle East, India, Southeast Asia, southern China, and occasionally the Mediterranean region.
People whose hemoglobin does not produce enough beta protein have beta thalassemia. It is found in people of Mediterranean descent, such as Italians and Greeks, and is also found in the Arabian Peninsula, Iran, Africa, Southeast Asia and southern China.
 Thalassemia is always inherited, passed on from parents to children through their genes. A child cannot develop the disease unless both parents carry the thalassemia gene.
Thalassemia is always inherited, passed on from parents to children through their genes. A child cannot develop the disease unless both parents carry the thalassemia gene.

The beta thalassemia syndromes are much more diverse than the alpha thalassemia syndromes due to the diversity of the mutations that produce the defects in the beta globin gene. Unlike the deletions that constitute most of the alpha thalassemia syndromes, beta thalassemias are caused by mutations on chromosome 11 that affect all aspects of beta globin production: transcription, translation, and the stability of the beta globin product. Most hematologists feel there are three general categories of beta thalassemia: beta thalassemia trait, beta thalassemia intermedia and beta thalassemia major.
Patients with Beta-Thalassemia Intermedia have varying effects from the disease - mild anemia might be their only symptom or they might require regular blood transfusions.
The most common complaint is fatigue or shortness of breath, heart palpitations (due to the anemia, and mild jaundice, which is caused by the destruction of abnormal red blood cells that result from the disease), bone abnormalities( Because the bone marrow is working overtime to make more red blood cells to counteract the anemia, children can experience enlargement of their cheek bones, foreheads, and other bones).
Beta thalassemia major was first described by a Detroit pediatrician, Thomas Cooley, in 1925. The child born with thalassemia major has two genes for beta thalassemia and no normal beta-chain gene. The child is homozygous for beta thalassemia. This causes a striking deficiency in beta chain production and in the production of Hb A. Thalassemia major is, therefore, a serious disease.
Anemia begins to develop within the first months after birth. It becomes progressively more and more severe. The infant fails to thrive (to grow normally) and often has problems feeding (due to easy fatigue from lack of oxygen, with the profound anemia), bouts of fever (due to infections to which the severe anemia predisposes the child) and diarrhea and other intestinal problems.
Treatment for Thalassemia :
Patients with thalassemia minor usually do not require any specific treatment. Treatment for patients with thalassemia major includes chronic transfusion therapy, iron chelation, splenectomy or removal of the spleen (rarely require splenectomy, although the development of bilirubin stones frequently leads to cholecystectomy), and Bone Marrow or Stem Cell Transplants also other name was called allogeneic hematopoietic transplantation (this is curative in some patients with thalassemia major).
Other Treatments :
People with severe thalassemia are more likely to get infections that can worsen their anemia. They should get an annual flu shot and the pneumonia vaccine to help prevent infections.
Folic acid is a B vitamin that helps build red blood cells. People with thalassemia should take folic acid supplements.
Researchers are also studying other treatments, such as gene therapy and fetal hemoglobin.
Diet for Patient Thalassemia :
Drinking tea may help to reduce iron absorption through the intestinal tract, Vitamin C may improve iron excretion in patients receiving iron chelation. Anecdotal reports suggest that large doses of vitamin C can cause fatal arrhythmias when administered without concomitant infusion of deferoxamine. Read more!
When there is not enough hemoglobin in the red blood cells, oxygen cannot get to all parts of the body. Organs then become starved for oxygen and are unable to function properly.
Hemoglobin contains two different kinds of protein chains named alpha and beta chains. Any deficiency in these chains causes abnormalities in the formation, size, and shape of red blood cells.
People whose hemoglobin does not produce enough alpha protein have alpha thalassemia. It is commonly found in Africa, the Middle East, India, Southeast Asia, southern China, and occasionally the Mediterranean region.
People whose hemoglobin does not produce enough beta protein have beta thalassemia. It is found in people of Mediterranean descent, such as Italians and Greeks, and is also found in the Arabian Peninsula, Iran, Africa, Southeast Asia and southern China.
Thalassemia is always inherited, passed on from parents to children through their genes. A child cannot develop the disease unless both parents carry the thalassemia gene.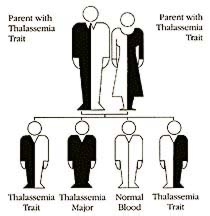
The beta thalassemia syndromes are much more diverse than the alpha thalassemia syndromes due to the diversity of the mutations that produce the defects in the beta globin gene. Unlike the deletions that constitute most of the alpha thalassemia syndromes, beta thalassemias are caused by mutations on chromosome 11 that affect all aspects of beta globin production: transcription, translation, and the stability of the beta globin product. Most hematologists feel there are three general categories of beta thalassemia: beta thalassemia trait, beta thalassemia intermedia and beta thalassemia major.
Patients with Beta-Thalassemia Intermedia have varying effects from the disease - mild anemia might be their only symptom or they might require regular blood transfusions.
The most common complaint is fatigue or shortness of breath, heart palpitations (due to the anemia, and mild jaundice, which is caused by the destruction of abnormal red blood cells that result from the disease), bone abnormalities( Because the bone marrow is working overtime to make more red blood cells to counteract the anemia, children can experience enlargement of their cheek bones, foreheads, and other bones).
Beta thalassemia major was first described by a Detroit pediatrician, Thomas Cooley, in 1925. The child born with thalassemia major has two genes for beta thalassemia and no normal beta-chain gene. The child is homozygous for beta thalassemia. This causes a striking deficiency in beta chain production and in the production of Hb A. Thalassemia major is, therefore, a serious disease.
Anemia begins to develop within the first months after birth. It becomes progressively more and more severe. The infant fails to thrive (to grow normally) and often has problems feeding (due to easy fatigue from lack of oxygen, with the profound anemia), bouts of fever (due to infections to which the severe anemia predisposes the child) and diarrhea and other intestinal problems.
Treatment for Thalassemia :
Patients with thalassemia minor usually do not require any specific treatment. Treatment for patients with thalassemia major includes chronic transfusion therapy, iron chelation, splenectomy or removal of the spleen (rarely require splenectomy, although the development of bilirubin stones frequently leads to cholecystectomy), and Bone Marrow or Stem Cell Transplants also other name was called allogeneic hematopoietic transplantation (this is curative in some patients with thalassemia major).
Other Treatments :
Diet for Patient Thalassemia :
Drinking tea may help to reduce iron absorption through the intestinal tract, Vitamin C may improve iron excretion in patients receiving iron chelation. Anecdotal reports suggest that large doses of vitamin C can cause fatal arrhythmias when administered without concomitant infusion of deferoxamine. Read more!
posted by Ocha at
12:50 AM
1 Comments
![]()
![]()